Data decyzji: 17 lutego 2026 r.
Organ: Główny Inspektor Farmaceutyczny (GIF)
Numer decyzji: 10/WC/ZW/2026
Sygnatura akt: NNJ.5453.15.2026.MRO.3
Główny Inspektor Farmaceutyczny wydał decyzję o wycofaniu z obrotu na terenie całego kraju serii H36032A produktu leczniczego Grofibrat 200 (Fenofibratum) 200 mg, kapsułki twarde. Przyczyną jest niezgodność wyników badań stabilności z wymaganiami specyfikacji jakościowej dla parametru uwalniania substancji czynnej.
Spis treści
Dane produktu
| Parametr | Wartość |
|---|---|
| Nazwa handlowa | Grofibrat 200 |
| Substancja czynna | Fenofibratum (fenofibrat) |
| Postać farmaceutyczna | Kapsułki twarde |
| Dawka | 200 mg |
| Wielkość opakowania | 30 kapsułek |
| GTIN | 05909990492114 |
| Nr pozwolenia na dopuszczenie do obrotu | 04921 |
| Podmiot odpowiedzialny | Gedeon Richter Polska Sp. z o.o., Grodzisk Mazowiecki |
| Wycofana seria | H36032A |
| Termin ważności serii | 30.06.2027 |
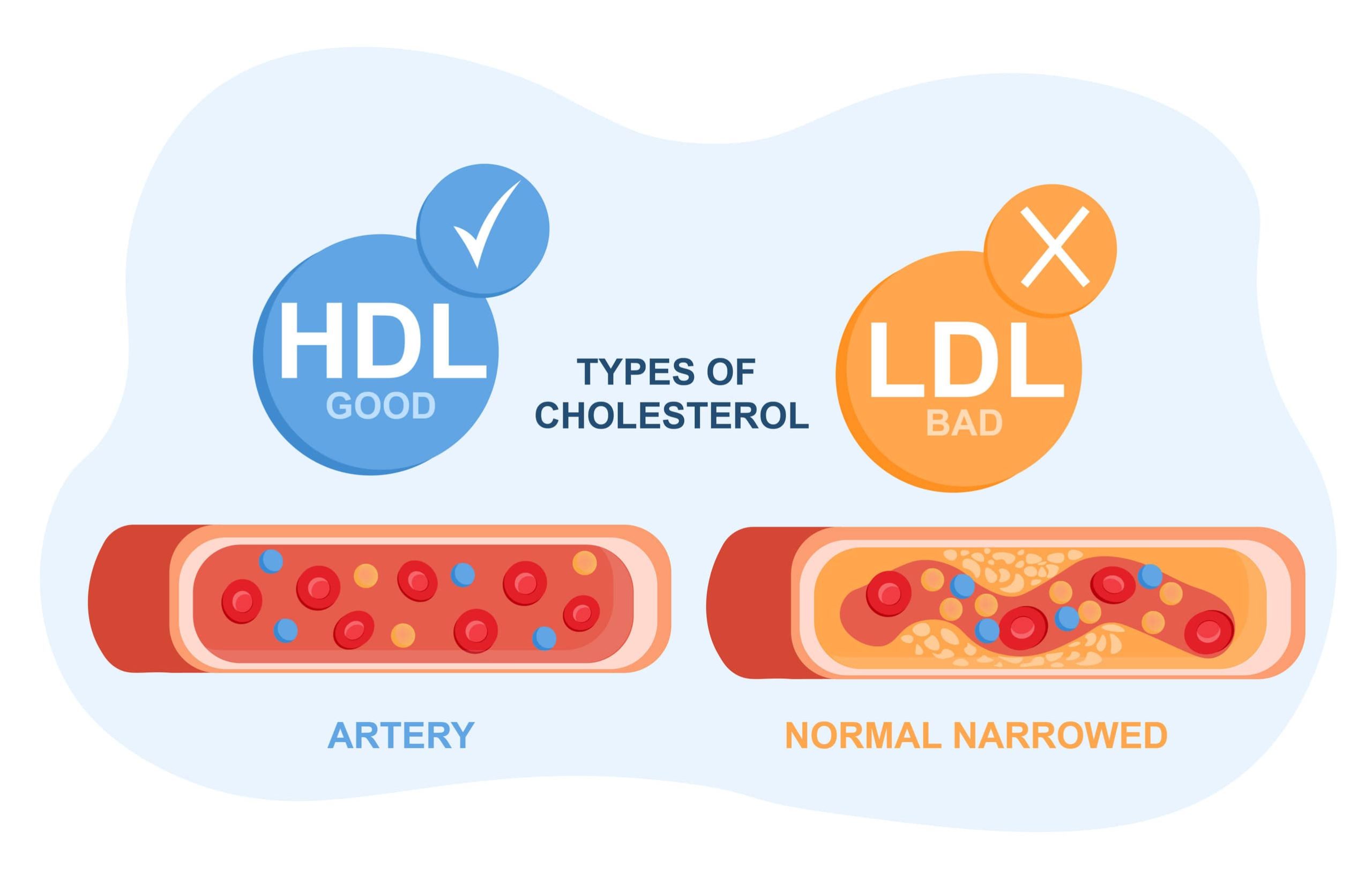
Co to jest Grofibrat 200? Opis leku
Substancja czynna — fenofibrat
Grofibrat 200 zawiera fenofibrat — substancję czynną z grupy fibratów, stosowaną w leczeniu zaburzeń lipidowych (dyslipidemii). Fenofibrat jest prolekiem, który po wchłonięciu ulega hydrolizie do aktywnej postaci — kwasu fenofibrowego.
Mechanizm działania
Fenofibrat działa poprzez aktywację receptorów jądrowych PPARα (receptor aktywowany przez proliferatory peroksysomów alfa). Prowadzi to do:
- obniżenia stężenia trójglicerydów — poprzez nasilenie lipolizy i zmniejszenie syntezy VLDL w wątrobie,
- podwyższenia stężenia HDL (tzw. dobrego cholesterolu) — przez zwiększenie ekspresji apolipoprotein A-I i A-II,
- obniżenia stężenia LDL — zwłaszcza frakcji małych, gęstych cząstek LDL, uznawanych za szczególnie aterogenne.
Wskazania do stosowania
Grofibrat 200 jest wskazany u dorosłych pacjentów w leczeniu:
- ciężkiej hipertriglicerydemii (izolowanego podwyższenia trójglicerydów),
- mieszanej dyslipidemii (współwystępowanie podwyższonego cholesterolu i trójglicerydów) — zazwyczaj jako uzupełnienie leczenia statynami, gdy kontrola samymi statynami jest niewystarczająca.
Dawkowanie i kontrola leczenia
Zgodnie z Charakterystyką Produktu Leczniczego zalecana dawka wynosi 1 kapsułkę 200 mg na dobę. Lek powinno się przyjmować podczas posiłku, co poprawia jego wchłanianie. Skuteczność terapii wymaga kontroli stężenia lipidów we krwi — jeśli po kilku miesiącach leczenia (zwykle trzech) nie uzyskano oczekiwanego efektu, lekarz powinien rozważyć zmianę lub rozszerzenie terapii.
Przyczyna wycofania — wynik OOS w badaniu stabilności
Co wykryto?
W długoterminowym badaniu stabilności produktu leczniczego dla serii H36032A stwierdzono wynik nieodpowiadający wymaganiom specyfikacji jakościowej (ang. Out of Specification, OOS) dla parametru uwalnianie substancji czynnej. Oznacza to, że fenofibrat uwalniał się z kapsułek poniżej dolnego progu określonego w zatwierdzonej dokumentacji rejestracyjnej.
Co zbadano w trakcie dochodzenia?
Wytwórca przeprowadził szczegółowe dochodzenie, w trakcie którego:
- wykluczono błąd analityczny jako przyczynę wyniku OOS,
- nie stwierdzono istotnych zmian porejestracyjnych (m.in. zmiany dostawcy substancji czynnej),
- nie odnotowano odchyleń w procesie wytwarzania — wszystkie krytyczne parametry były zgodne z wymaganiami,
- oceniono próbę archiwalną serii — wykazała ona zgodność ze specyfikacją, jednak z niższym wynikiem uwalniania niż w momencie zwolnienia serii,
- nie odnotowano żadnych reklamacji ze strony pacjentów ani zgłoszeń działań niepożądanych.
Ustalona przyczyna niezgodności
Na podstawie analizy porównawczej serii H36032A z seriami walidacyjną (H33098A) oraz H3B158A wytwórca wskazał, że niezgodność była wynikiem kumulacji dwóch czynników:
- większego udziału kapsułek bliskich dolnej granicy dopuszczalnej masy,
- niższej zawartości substancji czynnej (API) w kapsułkach.
W serii H36032A proces ustawień maszyny trwał dłużej niż w pozostałych seriach, co skutkowało wyprodukowaniem większej liczby kapsułek o niższej masie.
Działania naprawcze
Podmiot odpowiedzialny zaproponował działania korygujące w postaci zmiany dolnego limitu dopuszczalnych mas na sortownicy dla tego produktu.
Ocena ryzyka dla pacjenta
Podmiot odpowiedzialny przeprowadził medyczną ocenę ryzyka i stwierdził, że stwierdzone odchylenie nie powinno mieć wpływu na bezpieczeństwo stosowania produktu. Niemniej jednak GIF zwrócił uwagę na potencjalne ryzyko dla skuteczności terapii:
Obniżone uwalnianie substancji czynnej może wpłynąć na wyniki badań stężenia lipidów w surowicy, co z kolei może prowadzić do niepotrzebnej zmiany leczenia lub zastosowania dodatkowej farmakoterapii przez lekarza prowadzącego.
Z tego względu GIF uznał, że istnieje realne zagrożenie wynikające z możliwego braku zakładanej skuteczności leku i wydał decyzję o wycofaniu z obrotu oraz nadał jej rygor natychmiastowej wykonalności.
Podstawa prawna decyzji
Decyzja została wydana na podstawie:
- art. 122 ust. 1 oraz art. 108 ust. 4 pkt 2 ustawy z dnia 6 września 2001 r. – Prawo farmaceutyczne (Dz. U. z 2025 r. poz. 750 z późn. zm.),
- art. 104 § 1–2 i art. 108 § 1 ustawy z dnia 14 czerwca 1960 r. – Kodeks postępowania administracyjnego (Dz. U. z 2025 r. poz. 1691).
Informacje dla pacjentów
Co oznacza wycofanie dla pacjenta przyjmującego Grofibrat 200?
Jeżeli posiadasz lek z serii H36032A (data ważności: 30.06.2027), nie powinieneś go stosować. Numer serii znajduje się na opakowaniu — sprawdź go przed przyjęciem leku.
Co zrobić?
- Skontaktuj się ze swoim lekarzem lub farmaceutą — poinformuj o wycofaniu i zapytaj o możliwość otrzymania innej serii lub zamiennika.
- Nie przerywaj leczenia samodzielnie — fenofibrat jest stosowany w terapii przewlekłej i wymaga kontroli lekarskiej. Nagłe odstawienie leku bez konsultacji może być niekorzystne.
- Zwróć lek do apteki — wycofany produkt leczniczy podlega zniszczeniu; apteka powinna przyjąć go z powrotem.
Czy inne serie Grofibratu 200 są bezpieczne?
Decyzja GIF obejmuje wyłącznie serię H36032A. Pozostałe serie produktu nie są objęte wycofaniem.
Pouczenie — tryb odwoławczy
Od decyzji GIF nie przysługuje odwołanie w trybie standardowym. Podmiot odpowiedzialny (Gedeon Richter Polska Sp. z o.o.) może w terminie 14 dni od doręczenia decyzji złożyć wniosek o ponowne rozpatrzenie sprawy do Głównego Inspektora Farmaceutycznego. Decyzja podlega wykonaniu z chwilą doręczenia — złożenie wniosku o ponowne rozpatrzenie nie wstrzymuje jej wykonania.
Źródło: Decyzja nr 10/WC/ZW/2026 Głównego Inspektora Farmaceutycznego z dnia 17 lutego 2026 r., znak NNJ.5453.15.2026.MRO.3. Pełna treść dostępna na stronie gif.gov.pl.
{kind=link}
Niniejszy artykuł nie jest poradą medyczną i ma charakter wyłącznie informacyjny.